GMP Qualification Operations – Manufacturing of Biological Products
Analytical Instrument Qualification in compliance with USP <1058>
In 2008 the US Pharmacopeia (USP) implemented the general chapter <1058> on Analytical Instrument Qualification (AIQ) originated by the American Association of Pharmaceutical Scientists (AAPS). This chapter was updated in 2017 to propose an integrated approach to AIQ and Computerized System Validation (CSV) together with GAMP. The USP <1058> has become an important document since it is the only risk-based regulatory guidance regarding analytical instrument qualification (AIQ).

About Analytical Instruments
Analytical instruments and devices are essential for manufacturing and quality control of biopharmaceuticals. Most of these instruments link metrological and software-controlled functions. QA is responsible for assuring that their instruments are suitably qualified.
AIQ Definitions
The qualification of the analytical instruments verifies through documented evidence that the instrument operates within its operational and functional specifications as defined and specified.

Process Equipment Qualification in compliance with EU GMP Guide Annex 15
The manufacturing of biotechnological-derived medicinal products involves several process steps from upstream to downstream operations. Various process equipment is used for the bioprocessing of biopharmaceuticals and immunotherapies. The process equipment varies from standard to complex equipment and devices used for production.
Equipment used in a GMP-related environment should be suitable and should not present any hazard to the product. The location and installation of the equipment should be adequate to minimize risk and contamination.
Objective
The objective of process equipment qualification is to verify the fitness for purpose for its intended use and to ensure that process equipment meet their specifications as devised in the URS.
Definitions
Process equipment qualification is the verification that the equipment is properly designed, installed and operates within specified specifications and their associated acceptance criteria.
Integrated Qualification and Computerized System Validation (CSV)
Most analytical instruments and process equipment are combined with computerized systems and contribute significantly to datasets. Software must be categorized and specified, computerized systems must be specified and both must be validated. GMP-regulated laboratories require data to be accurate, complete, reliable and consistent.
Commissioning and Qualification
Commissioning & Qualification (C&Q) applies Good Engineering Practice (GEP) into design and construction and GMP qualification principles to assembly, installation, testing and documentation to manufacturing systems.
C&Q combines Good Engineering Practice (GEP), the “Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment (ASTM E2500)”, and GMP qualification principles, to establish commissioning and qualification into design, construction, assembly, installation, testing and documentation to manufacturing systems.
Commissioning & Qualification (C&Q) focusses on the identification on critical design elements (CDEs) in terms of critical quality attributes (CQAs) and critical process parameters (CPPs) regarding product quality and safety.

Main C&Q Phases
- Design Review and Design Qualification
- C&Q Testing and Documentation
- C&Q Acceptance and Release
Main C&Q Requirements
- URS
- System Classification
- System Risk Assessment
System Risk Assessment
- Identify system risks in terms of product and process quality
- Evaluate and analyze the identified risks
- Define risk controls in terms of CDEs (critical design elements)
- Define system alarms
- Link testing activities to CQAs and CPPs
Process Facility
Process line and process facility qualification (e.g. Sterile Fill Line Equipment) for sterile pharmaceutical manufacturing is one of the most critical assets. For aseptic manufacturing in compliance with cGMP, EU GMP Guide Annex 1, FDA guidance, and ISO 14644 principles are applicable.

GMP Regulations and Requirements
- EU GMP Guide Annex 11
- EU GMP Guide Annex 15
- FDA 21 CFR Part 11
- GAMP 5
- FDA, EMA, EU, MHRA
- WHO GMP
Summary
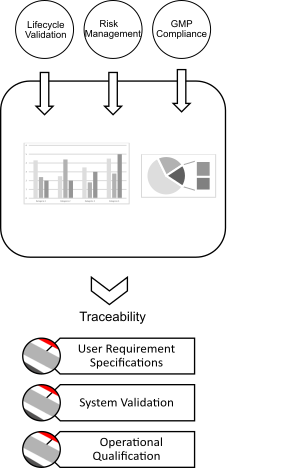
The use of qualified instruments is mandatory, as it is required by regulatory authorities such as the U.S. Food & Drug Administration (FDA) or European Medicines Agency (EMA) to receive the manufacturing authorization.
Qualified instruments contribute to client’s confidence that the analytical instruments and process equipment used for quality control and manufacturing perform suitably according to their intended purpose.
Analytical instruments should be qualified, calibrated, inspected and maintained at defined intervals to ensure adequate performance. The Guideline for Qualification and Validations are stated in the EU GMP Guide Annex 15.
The URS and the risk assessment are the most important documents as they impact directly the outcome of the qualification. However, user requirements and system specifications are not necessarily defined equally between supplier and laboratory. The devise of a thorough URS is of utmost avail and influences the outcome of the qualification, as the URS is linked to the operational qualification (OQ).